User Guide
You need coordinate files for each type of molecule you want your simulation to have. For example, if you are going to simulate a solution of water and ions, you will need a coordinate file for one water molecule, and independent coordinate files for each of the ions. These coordinate files may be in the PDB, TINKER, or XYZ (MOLDEN) formats.
Of course, you also need the Packmol package, which you can get from:
http://m3g.iqm.unicamp.br/packmol
by clicking on the Download link.
You can download the file packmol-21.0.0.tar.gz which contains the
whole source code of Packmol. The 21.0.0 is the version number, which
might change relative to this example.
Once you have downloaded the packmol-21.0.0.tar.gz file from the home page,
you need to expand the files and compile the package. This is done by:
Expanding the files:
tar -xvzf packmol-21.0.0.tar.gz
This will create a directory called packmol inside which you can find
the source code. You can build the executable by:
cd packmol
make
That's it! If no error was reported, the packmol executable was built.
Note that 21.0.0 is the version number, which may differ relative to this example.
If you have problems:
Let the configure script find a suitable compiler for you:
chmod +x ./configure # Makes the script executable
./configure # Executes the scriptIf the script was not able to find a suitable compiler, then you can manually set the compiler by:
./configure /path/to/your/compiler/yourcompilerThen, run the "make" command again:
makeIf no error was detected, an executable called packmol is now ready.
From the command line, there are two alternatives to run Packmol:
Option 1: Using input file redirection
packmol < packmol.inpOption 2: Using command line arguments (from v21.1.0 onwards)
packmol -i packmol.inp -o output.pdb
The -o output.pdb is optional, and will overwrite the definition of the
output file in the input file.
The packmol.inp file is the input file (you can obtain example files by
clicking at the 'Input examples' link on the left).
A successful packing will end with something like:
------------------------------
Success!
Final objective function value: .22503E-01
Maximum violation of target distance: 0.000000
Maximum violation of the constraints: .78985E-02
------------------------------Where the maximum violation of the target distance indicates the difference between the minimum distance between atoms required by the user and that of the solution. It will not be greater than 10-2. The maximum violation of the constraints must not be greater than 10-2.
Common issues:
-
If you get "Command not found" when running Packmol, use:
(with a "./" before "packmol") or add the directory where the packmol executable is located to your path../packmol < packmol.inp - If you run packmol and get the message "Killed", this is because the package is trying to allocate more memory than available for static storage. Open the "sizes.i" file and decrease the "maxatom" parameter to the number of atoms of your system, compile the package again, and try again.
The minimal input file must contain the distance tolerance required (for systems at room temperature and pressure and coordinates in Angstroms, 2.0 Å is a good value [note]). This is specified with:
tolerance 2.0The file must also contain the name of the output file to be created:
output test.pdband the file type (pdb, tinker, or xyz; pdb is the default value):
filetype pdb
At least one type of molecule must be present. This is set by the
structure ... end structure section. For example, if
water.pdb is the file containing the coordinates of a single water molecule,
you could add to your input file something like:
structure water.pdb
number 2000
inside cube 0. 0. 0. 40.
end structure
This section specifies that 2000 molecules of the water.pdb type will
be placed inside a cube with minimum coordinates $(x,y,z) = (0,0,0)$ and
maximum coordinates $(40,40,40)$. Therefore, this minimum input file must be:
tolerance 2.0
output test.pdb
filetype pdb
structure water.pdb
number 2000
inside cube 0. 0. 0. 40.
end structureRunning Packmol with this input file will fill a cube of side 40.0 Å with 2000 water molecules. Every pair of atoms of different molecules will be separated by at least 2.0 Å and the molecules will be randomly distributed inside the cube.
You can add more types of molecules to the same region, or to different
regions of the space, simply by adding other structure ... end structure
sections to the input file.
The coordinate file of a single molecule contains, for example, 10
atoms. You can restrain a part of the molecule to be in a specified
region of the space. This is useful for building vesicles where the
hydrophilic part of the surfactants must be pointing to the aqueous
environment, for example. For the 10 atoms molecule, this is done by
using the keyword atoms, as in:
structure molecule.pdb
inside cube 0. 0. 0. 20.
atoms 9 10
inside box 0. 0. 15. 20. 20. 20.
end atoms
end structureIn this case, all the atoms of the molecule will be put inside the defined cube, but atoms 9 and 10 will be restrained to be inside the box.
There are several types of constraints that can be applied both to whole molecules or to parts of the molecule. These constraints define the region of the space in which the molecules must be at the solution. Very ordered systems can be built in such a way. The constraints are:
fixed x y z a b g
This options holds the molecule
fixed in the position specified by the parameters. x, y, z, a, b, g,
which are six real numbers. The first three determine the translation of
the molecule relative to its position in the coordinate file. The latter
three parameters are rotation angles (in radians). For this option it is
required that only one molecule is set. It may be accompanied by the
keyword center. If this keyword is present the first three numbers
are the position of the barycenter (not truly the center of mass,
because we assume that all atoms have the same mass). Therefore this
keyword must be used in the following context:
structure molecule.pdb
number 1
center
fixed 0. 0. 0. 0. 0. 0.
end structureIn this example, the molecule will be fixed with its center at the origin and no rotation.
inside cube xmin ymin zmin d
xmin, ymin, zmin and d are four real numbers. The coordinates (x,y,z) of the atoms restrained by this option will satisfy, at the solution:
ymin < y < ymin + d
zmin < z < zmin + d
outside cube xmin ymin zmin d
xmin, ymin, zmin and d are four real numbers. The coordinates (x,y,z) of the atoms restrained by this option will satisfy, at the solution:
y < ymin or y > ymin + d
z < zmin or z > zmin + d
inside box xmin ymin zmin xmax ymax zmax
xmin, ymin, zmin, xmax, ymax and zmax are six real numbers. The coordinates (x,y,z) of the atoms restrained by this option will satisfy, at the solution:
ymin < y < ymax
zmin < z < zmax
outside box xmin ymin zmin xmax ymax zmax
xmin, ymin, zmin, xmax, ymax and zmax are six real numbers. The coordinates (x,y,z) of the atoms restrained by this option will satisfy, at the solution:
y < ymin or y > ymax
z < zmin or z > zmax
Spheres are defined by equations of the general form
$$ (x - a)^2 + (y - b)^2 + (z - c)^2 - d^2 = 0 $$and, therefore, you must provide four real parameters a, b, c and d in order to define it. The input syntax is, for example:
inside sphere 2.30 3.40 4.50 8.0and therefore the coordinates of the atoms will satisfy the equation
$$ (x - 2.30)^2 + (y - 3.40)^2 + (z - 4.50)^2 - 8.0^2 \leq 0.0 $$Other input alternative would be:
outside sphere 2.30 3.40 4.50 8.0
The outside parameter is similar to the inside parameter, but the
equation above uses $\geq$ instead of $\leq$ and, therefore, the
atoms will be placed outside the defined sphere.
Ellipsoids are defined by the general equation
$$ \frac{(x-a_1)^2}{a_2^2} + \frac{(y-b_1)^2}{b_2^2} + \frac{(z-c_1)^2}{c_2^2} - d^2 = 0 $$The parameters must be given as in the sphere example, but now they are 7, and must be entered in the following order:
inside ellipsoid a1 b1 c1 a2 b2 c2 d
The coordinates (a1,b1,c1) will define the center of the ellipsoid, the coordinates (a2,b2,c2) will define the relative size of the axes and d will define the volume of the ellipsoid. Of course, the command:
outside ellipsoid a1 b1 c1 a2 b2 c2 d
can also be used in the same manner as the parameters for spheres. Note that the case a2 = b2 = c2 = 1.0 provides exactly the same as the sphere parameter. The parameters for the ellipsoid are not normalized. Therefore, if a2, b2 and c2 are large, the ellipsoid will be large, even for a small d.
The planes are defined by the general equation
$$ ax + by + cz - d = 0 $$And it is possible to restrict atoms to be above or below the plane. The syntax is:
above plane 2.5 3.2 1.2 6.2
below plane 2.5 3.2 1.2 6.2
where the above keyword will make the atoms satisfy the condition
and the below keyword will make the atoms satisfy
Note: the above notation was introduced in version 20.1.1 (Sept. 5, 2020)
In order to define a cylinder, it is necessary first to define a line oriented in space. This line is defined in Packmol by the parametric equation
$$ p = (a_1, b_1, c_1) + t \times (a_2, b_2, c_2) $$where $t$ is the independent parameter. The vector $(a_2, b_2, c_2)$ defines the direction of the line. The cylinder is therefore defined by the distance to this line, $d$, and a length $l$. Therefore, the usage must be:
inside cylinder a₁ b₁ c₁ a₂ b₂ c₂ d l
outside cylinder a₁ b₁ c₁ a₂ b₂ c₂ d lHere, the first three parameters define the point where the cylinder starts, and l defines the length of the cylinder. d defines the radius of the cylinder. The simpler example is a cylinder oriented in the x axis and starting at the origin, such as:
inside cylinder 0. 0. 0. 1. 0. 0. 10. 20.This cylinder is specified by the points that have a distance of 10. to the x axis (the cylinder has a radius of 10.). Furthermore, it starts at the origin, therefore no atom restricted by this cylinder will have an x coordinate less than 0. Furthermore, it has a length of 20. and, as such, no atom will have an x coordinate greater than 20. The orientation of the cylinder, parallel to the x axis is defined by the director vector (1,0,0), the fourth, fifth and sixth parameters. Cylinders can be oriented in any direction in space.
It is possible to constrain rotations of all molecules of each type, so that they have some average orientation in space.
The keywords to be used are, within a structure...end structure section:
constrain_rotation x 180. 20.
constrain_rotation y 180. 20.
constrain_rotation z 180. 20.
Each of these keywords restricts the possible rotation angles around
each axis to be within 180±20 degrees (or any other value). For a
technical reason the rotation around the z axis will, alone, be
identical to the rotation around the y axis (we hope to fix
this some day). Constraining the three rotations will constrain
completely the rotations. Note that to have your molecules oriented
parallel to an axis, you need to constrain the rotations relative to the
other two.
For example, to constrain the rotation of your molecule along the
z axis, use something like:
constrain_rotation x 0. 20.
constrain_rotation y 0. 20.
Note that these rotations are defined relative to the molecule in the
orientation which was provided by the user, in the input PDB file.
Therefore, it is a good idea to orient the molecule in a reasonable way
in order to understand the rotations. For example, if the molecule is
elongated in one direction, a good idea is to provide the molecule with
the larger dimension oriented along the z axis.
(gaussian surface)
Gaussian surfaces are defined by the equation
$$ h \times exp\left[−\frac{(x-a_1)^2}{2a_2^2}−\frac{(y-b_1)^2}{(2b_2^2)}\right]-(z-c)=0 $$Parameters $(a_1, b_1)$ define center of the gaussian, while $c$ specifies the height in the $z$ dimension. $a_2$ and $b_2$ set the width of the gaussian in $x$ and $y$, respectively, while $h$ specifies its height. It is possible to restrict atoms to be over or below the gaussian plane. The gaussian surface as implemented is restricted to be over the $xy$ plane. The syntax is:
over xygauss a₁ b₁ a₂ b₂ c h
below xygauss a₁ b₁ a₂ b₂ c hFor example, using:
over xygauss 21.0 5.0 10.0 20.0 -23.0 15.0the atoms satisfy the condition
$$ 15.0 \times exp\left[−\frac{(x-21.0)^2}{2(10.0)^2}−\frac{(y-5.0)^2}{2(20.0)^2}\right]-(z+23.0) \geq 0.0 $$As of version 20.15.0, Packmol supports Orthorhombic periodic boundary conditions (thanks to Yi Yao for the contribution).
Usage:
For example, to set a periodic boundary with a box of 30x30x60 Angstroms, use:
pbc 30.0 30.0 60.0
In this case, all molecules will be set to stay in the box defined by minimum and maximum
coordinates 0.0 0.0 0.0 30.0 30.0 60.0, but with periodic boundary
conditions at the boundaries for the computation of the inter-atomic distances.
And if six parameters are provided, the periodic boundaries will be applied at the minimum and maximum coordinates provided:
pbc -15.0 -15.0 -30.0 15.0 15.0 60.0This keyword is a global keyword, and will affect all but fixed molecules. For example, this creates a periodic box of water:
tolerance 2.0
output water_box.pdb
pbc 30.0 30.0 60.0
structure water.pdb
number 1000
endAnd this will create an interface of water and carbon tetrachloride, with the interface at the (z=0) plane:
tolerance 2.0
output interface.pdb
pbc -20.0 -20.0 -30.0 20.0 20.0 30.0
structure water.pdb
number 1000
below plane 0.0 0.0 1.0 0.0
end structure
structure ccl4.pdb
number 200
above plane 0.0 0.0 1.0 0.0
end structureImportantly, the positions of the constraints refer to the periodic reference box, which is defined by the minimum and maximum coordinates provided. Fixed molecules (proteins, or other macromolecules) must fit inside the the periodic box after positioning.
It is possible (from version 15.133 on) to attribute different radii to
different atoms during the packing procedure. The default behavior is
that all atoms will be distant to each other at the final structure at least
the distance defined by the tolerance parameter. In this
case it is possible to think that the radius of every atom is half the
distance tolerance. A tolerance of 2.0 Angs is usually fine for
simulation of molecular systems using all-atom models.
Most times, therefore, you won't need this option. This was requested by users that want to pack multiscale models, in which all-atom and coarse-grained representations are combined in the same system.
It is easy to define different radii to different molecules, or to atoms
within a molecule. Just add the "radius" keyword within the
structure ... end structure section of a molecule type, or
within atoms ... end atoms section of an atom selection.
For example, in this case:
tolerance 2.0
structure water.pdb
number 500
inside box 0. 0. 0. 30. 30. 30.
radius 1.5
end structurethe radius of the atoms of the water molecules will be 1.5. Note that this implies a distance tolerance of 3.0 within water molecules.
In this case, on the other side:
tolerance 2.0
structure water.pdb
number 500
inside box 0. 0. 0. 30. 30. 30.
atoms 1 2
radius 1.5
end atoms
end structureonly atoms 1 and 2 of the water molecule (as listed in the water.pdb file) will have a radius of 1.5, while atom 3 will have a radius of 1.0, as defined by the tolerance of 2.0
Always remember that the distance tolerance is the sum of the radii of each pair of atoms, and that the greatest the radii the harder the packing. Also, keep in mind that your minimization and equilibration will take care of the actual atom radii, and Packmol is designed only to give a first coordinate file.
Finally, currently the restrictions are set to be fulfilled by the center of the atoms. Therefore, if you are using large radii, you might want to adjust the sizes of the boxes, spheres, etc., so that the whole atoms are within the desired regions. For standard all-atom simulations this is not usually an issue because the radii are small.
The Packmol distribution includes the solvate.tcl script,
which is used to solvate large molecules, usually proteins, with water
and ions (Na+ and Cl-). Given the PDB file of the
biomolecule, just run the script with:
solvate.tcl PROTEIN.pdb
And the script will create a input file for packmol called
packmol_input.inp. With this file, run Packmol with
packmol < packmol_input.inpAnd your large molecule will be solvated by a shell of 15. Angs. of water, and ions to keep the system neutral and a physiological NaCl concentration of 0.16M. The script usually makes reasonable choices for every parameter (number of water molecules, number of ions, etc.), but these may be controlled manually with additional options, as described below:
solvate.tcl structure.pdb -shell 15. -charge +5 -density 1.0 -i pack.inp
-o solvated.pdb| Where: |
structure.pdb is the pdb file to be solvated (usually a protein)
" 15." is the size of the solvation shell. This
is an optional parameter. If not set, 15. will be used.
+5 is the total charge of the system, to be neutralized.
This is also and optional parameter, if not used, the package
considers histidine residues as neutral, Arg and Lys as +1
and Glu and Asp as -1. The Na+ and Cl- concentrations are set
the closest possible to 0.16M, approximately the physiological
concentration.
Alternatively, use the -noions to not add any ions, just water.
1.0 is the desired density. Optional. If not set, the density
will be set to 1.0 g/ml.
solvated.pdb: is the (optional) name for the solvated system
output file. If this argument is not provided, it will be the default
solvated.pdb file.
pack.inp: is the (optional) name for the packmol input file that
will be generated. If not provided, packmol_input.inp will be used.
|
All these options are output when running the "solvate.tcl" script without any parameter. The script also outputs the size of the box and the suggested periodic boundary condition dimensions to be used.
Since Packmol will create one or more copies of your molecules in a
new PDB file, there are some options on how residue numbers are set to
these new molecules. There are four options, which are set with the
resnumbers keyword. This keyword may assume three values, 0, 1,
2 or 3, and must be inserted within the structure ... end structure
section of each type of molecule. The options are:
resnumbers 0
In this case the residue numbers of all residues will correspond to the
molecule of each type, independently of the residue
numbering of the original pdb file. This means that if you pack 10
water molecules and 10 ethanol molecules, the water molecules will be numbered
1 to 10, and the ethanol molecules will be numbered 1 to 10.
resnumbers 1
In this case, the residue numbers of the original pdb files are kept unmodified. This means that if you pack 10 proteins of 5 residues, the residue numbers will be preserved and, therefore, they will be repeated for equivalent residues in each molecule of the same protein.
resnumbers 2
In this case, the residue numbers of all residues for this structure will be numbered sequentially according to the number of residues that are printed previously in the same file. This means that if you pack 10 proteins of 5 residues, there will be residue numbers ranging from 1 to 50.
resnumbers 3
In this case, the numbering of the residues will correspond to the sequential numbering of all residues in the file. That is, if you pack a protein with 150 residues followed by 10 water molecules, and use this option for the water molecules, the water molecules will be numbered from 151 to 161. Note: If you use this option for a protein, the whole protein will have its residue numbers overwritten by the same number, corresponding to the residue that would follow in the complete structure.
For example, this keyword may be used as in:
structure peptide.pdb
number 10
resnumbers 1
inside box 0. 0. 0. 20. 20. 20.
end structureDefault: The default behavior is to use 0 for structures with only one residue and 1 for structures with more than one residue.
Chain identifier:
It is also possible to modify the "chain" identifiers of PDB files.
By default, each type of molecule is set to a "chain". On the other side,
using the changechains keyword within the structure...end structure
section of a type of molecule, the chains will alternate between
two values ("A" and "B" for example). This might be useful if the
molecules are peptides, and topology builders sometimes think
that the peptides of the same chain must be joint by covalent bonds.
This is avoided by alternating the chain from molecule to molecule.
Additionally each structure can have a specific chain identifier, set
by the user with the option: chain D
where "D" is the desired identifier (Do not use "#").
From version 16.143 on, it is possible to build the system from multiple and independent executions of Packmol by the use of restart files. In order to write a restart file, the following keyword must be used:
restart_to restart.pack
where restart.pack is the name of the restart file to be
created. It is possible to write restart files for the whole system, if
the keyword is put outside structure...end structure sections,
or to write a restart file for a specific part of the system, using, for
instance:
structure water.pdb
number 1000
inside cube 0. 0. 0. 40.
restart_to water1.pack
end structureThis will generate a restart file for the water molecules only.
These restart files can be used to start a new execution of Packmol with
more molecules. The restart_from keyword must then be used. For
example:
structure water.pdb
number 1000
inside cube 0. 0. 0. 40.
restart_from water1.pack
end structure
The new input file might contain other molecules, as a regular Packmol input file, and these water molecules will be packed together with the new molecules, but starting from the previous runs. This can be used, for example, to build solvated bilayers by parts. For instance, the bilayers could be built and, later, different solvents can be added to the bilayer, without having to restart the whole system construction from scratch every time. This could also be used to add some molecule to the bilayer.
Tip: the restart file can be used to restart the position of a
smaller number of molecules of the same type.
For instance, if a new molecule is introduced
inside a previous set of molecules (a lipid bilayer, for instance), you
can tell Packmol to pack less molecules of the original set, in order to
provide space for the new structure, while using the original restart
file of more molecules. That is, a restart_from water1.pack
similar to the ones of the example above could be used to restart the
positions of 800 molecules.
Sometimes Packmol is not able to find an adequate packing solution. Here are some tips to try to overcome these difficulties:
| • Look at the best solution obtained, many times it is good enough to be used. | ||||||||||
| • Simulate the same problem with only a few molecules of each type. For example, instead of using 20 thousand water molecules, put 300, and see if they are in the correct regions. | ||||||||||
| • If you have large molecules, try running the program twice, one to pack these molecules, and then use the solution as fixed molecule for the next packing, in which solvation is included. This may be particularly useful for building solvated membranes. Build the membrane first and then use it as a fixed molecule for a solvation run. | ||||||||||
• You can change some options of the packing procedure to try
improve the optimization:
|
There are some input options which are probably not of interest of the general user, but may be useful in specific contexts. These keywords may be added in the input file in any position.
Control of PDB CONECT lines parsing:
Two keywords control what should be done with the information in CONECT lines of PDB files:
Usage: ignore_conect
Most times the connectivity of PDB files should be ignored, because
the correct connectivity is defined with topology files that are specific for the simulation
package to be used. Use this keyword if the parsing of the CONECT lines is causing any issue.
Usage: non_standard_conect
By default the CONECT fields of PDB files are fixed-width, and support only atom indices as
large as 99999. If this keyword is used, then the indices are expected to be separated by
spaces, and there is no limit for the printed integers. But this does not conform with the
standard PDB file format.
By default, both ignore_conect and non_standard_conect are set to false,
thus packmol will expect properly formatted CONECT lines.
Write a CHARMM CRD file as output:
Usage: writecrd output.crd
The segment identifier for each type of molecule can be set by adding,
within the structure ... end structure section of each type,
the following keyword: segid NAME.
Add the TER flag between every molecule (AMBER uses this):
Usage: add_amber_ter
and
Usage: amber_ter_preserve
(this option will preserve TER flags within input structures, fixed or not from v20.14.0 on).
Add box side information to output PDB File (GROMACS uses this):
Usage: add_box_sides
If no PBCs are used, this keyword adds a CRYST1 field to the output PDB file,
where the length of the unitcell in each direction will be the maximum and
minimum coordinates of the atoms in the solution added by 1.1*tolerance.
This can be used to prevent clashes at the boundaries. Nevertheless, it is
recommended to explicit use PBCs with the pbc keyword.
Display, or not, connectivity of molecules in output PDB files:
Usage: connect yes/no
By default, if the input PDB file of a molecule has CONECT fields,
the output PDB File of packmol will contain also CONECT fields
with the appropriate connectivity of the corresponding molecules. If you
do not want this, add connect no to the structure section of
this molecule, and the output of the connectivity will be suppressed.
Increase maximum system dimensions:
Usage: sidemax [real] (ex: sidemax 2000.d0)
sidemax is used to build an initial approximation of the molecular
distribution. Increase sidemax if your system is very large, or your
system may look cut out by this maximum dimension. All system
coordinates must fit within -sidemax and +sidemax, and
using a sidemax
that approximately fits your system may accelerate a little bit the
packing calculation. The default value is 1000.d0.
Change random number generator seed:
Usage: seed [integer] (ex: seed 191917)
Use seed -1 to generate a seed automatically from the computer
time.
Use a truly random initial point for the minimization:
(the default option is to generate a roughly homogeneous-density initial approximation)
Usage: randominitialpoint
Avoid, or not, overlap with fixed molecules at initial point:
(avoiding this overlaps is generally recommended, but sometimes generates gaps that are too large)
Usage: avoid_overlap yes/no
Change the maximum number of Gencan iterations per loop:
Usage: maxit [integer]
Change the maximum number of optimization loops:
(if this option is used inside the structure section, it affects the number of optimization
loops only of that specific structure)
Usage: nloop [integer]
Change the precision required for the solution:
How close the solution must be to the desired distances to be considered correct.
Usage: precision [real] (Default: 0.01)
Change the frequency of output file writing:
Usage: writeout [integer]
Write the current point to output file even if worse than best point:
(used for checking purposes only)
Usage: writebad
Change the optimization subroutine printing output:
Usage: iprint1 [integer] and/or iprint2 [integer]
where the integer must be 0, 1, 2 or 3.
Change the number of bins of the linked-cell method (technical):
Usage: fbins [real]
The default value is the square root of three.
Write all atom and residue indices in hexadecimal format (v20.16.0):
Usage: hexadecimal_indices
Compare analytical and finite-difference gradients:
This is only for testing purposes. Writes chkgrad.log file containing the comparison.
Usage: chkgrad
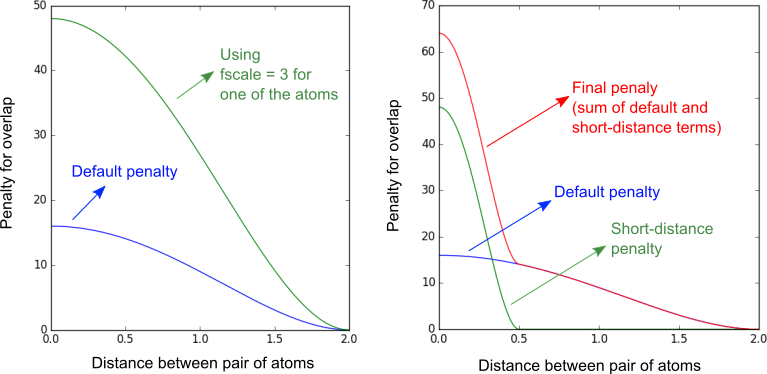
Options to change the shape of the minimum-distance penalty function
Usage: fscale [real]
This option must be used within structure ... end structure or
atoms ... end atoms sections, to change the weight of the
penalty function, as illustrated in the images below. The fscale defines
how greater the penalty is relative to the default penalty.
Usage: use_short_tol
Usage: short_tol_dist [real]
Usage: short_tol_scale [real]
These options define a new penalty tolerance for short distances, to increase the
repulsion between atoms when they are too close to each other. The "use"
parameter turns on this feature, using default parameters (dist=tolerance/2; scale=3).
The "dist" parameter defines from which distance the penalty is applied, and
the "scale" parameter how greater this penalty is relative to the
default penalty. They affect the penalty of all atoms. Remember that the
"tolerance" is twice the size of the "radius" of each atom. The figure below, on the
right, illustrates for example the build up of the penalty function using
short_tol_dist=0.5; short_tol_scale=3, with the default overall tolerance
of 2.0 set.
Usage: short_radius [real]
Usage: short_radius_scale [real]
These parameters define a new penalty for short distances, but for specific
atoms. These options must be used within structure ... end structure or
atoms ... end atoms sections. Note that, in this case, the
radius is defined (the actual tolerance is the sum of the radii of each
pair of atoms; for atoms without an explicit radius definition, the radius
is one half of the overall set tolerance parameter).
Here are some tutorials developed by third-parties which might be useful:
-
Windows: Tutorial - Using Packmol to generate solvation shell around molecules
by: Filipe Camargo Dalmatti Alves Lima -
PACKMOL tutorial- Solvation of proteins in non-standard solvents
by: Mohamed Shehata -
Preparation of initial configuration of polymers for MD simulation
by: Jeremy Wong