Protein conformational changes
Consider these two states of a model protein, a native and a denatured (straight chain) state, obtained from a simulation. Here the conformational change could be of any kind. We load the structures of the two states:
using PDBTools
native_state = read_pdb(PDBTools.MJC_NATIVE, "protein")
desnat_state = read_pdb(PDBTools.MJC_DESNAT, "protein") Vector{Atom{Nothing}} with 999 atoms with fields:
index name resname chain resnum residue x y z occup beta model segname index_pdb
1 N SER A 2 1 14.210 24.950 39.381 0.80 0.00 1 AP1 37
2 HN SER A 2 1 13.353 24.515 39.657 0.80 0.00 1 AP1 38
⋮
998 C LEU A 70 69 -26.425 41.129 -200.385 0.80 0.00 1 AP2 1034
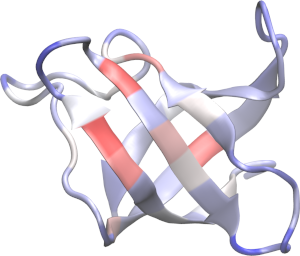
999 O LEU A 70 69 -27.110 41.780 -201.182 0.80 0.00 1 AP2 1035The denatured state has a greater surface area than the native state. Thus, cosolvents that bind preferentially to the surface, as urea, should promote a stabilization of the denatured state. This is obtained with:
m = mvalue(native_state, desnat_state, "urea"; model=MoeserHorinek)MValue{MoeserHorinek} - 69 residues - cosolvent: "urea"
Total m-value: -1.2025213 kcal mol⁻¹
Backbone contributions: -0.675961 kcal mol⁻¹
Side-chain contributions: -0.5265603 kcal mol⁻¹Where the tot, bb and sc fields contain, respectively, the total, backbone and side-chain contributions. The MValue object contains, additionally, the contribution of the side chain and backbone of each amino acid residue type for the m-value, in the residue_contributions_bb and residue_contributions_sc fields.
We can set the beta fields (for example) of the atoms as the residue contributions:
for (ir, r) in enumerate(eachresidue(native_state)) # iterate over residues
# total contribution of residue ir
c_residue = m.residue_contributions_sc[ir] + m.residue_contributions_bb[ir]
for at in r # iterate over atoms in residue
at.beta = c_residue
end
end
write_pdb("contrib.pdb", native_state)And with that get an image (here produced with VMD) of the contributions of the residues to the transfer free energies: