Simulação de Dinâmica Molecular Microcanônica
Abra o arquivo md-simple.jl, que contém o algoritmo de simulação. A simulação começa com velocidades aleatórias, ajustadas para a média termodinâmica desejada de 0.6 unidades/átomo ($kT=0.6$ em um sistema bidimensional). A essa energia cinética média chamaremos de "temperatura". O algoritmo de integração é Velocity-Verlet, que consiste em propagar as posições com
\[\vec{x}(t+\Delta t) = \vec{x}(t) + \vec{v}(t)\Delta t + \frac{1}{2}\vec{a}(t)\Delta t^2\]
sendo $\vec{a}(t)=\vec{F}(t)/m$, onde $\vec{F}(t)$ é a força no tempo corrente. A força em seguida é calculada em um tempo posterior com
\[\vec{F}(t+\Delta t) = -\nabla V\left[\vec{x}(t)\right]\]
e então as velocidades no instante seguinte são calculadas com
\[\vec{v}(t+\Delta t) = \vec{v}(t) + \frac{1}{2}\left[ \frac{\vec{F}(t)}{m}+\frac{\vec{F}(t+\Delta t)}{m}\right]\]
completando o ciclo. Neste exemplo as massas são consideradas unitárias, para simplificar. A simulação é executada por nsteps passos, com passo de integração $\Delta t$, este sendo um parâmetro de entrada, dt, definido em Options.
A simulação não tem controle de temperatura ou de pressão. É uma propagação da trajetória segundo as leis de Newton, que deveriam conservar a energia. A isso se chama uma simulação "microcanônica", ou "NVE" (que conserva, em princípio, o número de partículas, o volume e a energia total).
3.1. Passo de integração
Para realizar uma MD simples, com um passo de integração de dt=1.0, execute o comando:
julia> out = md(sys,Options(dt=0.1));Em princípio, está previsto realizar 2000 passos de integração das equações de movimento. Teste passos de integração entre 1.0 e 0.01. Veja o que acontece com a energia. Veja o que acontece com a energia cinética média, que foi inicializada em 0.6 unidades/átomo. Discuta a escolha do passo de integração, e os valores de energia cinética obtidos. As simulações seguintes vão usar um passo de integração dt = 0.05.
É possível controlar a frequência de impressão e o número de pontos salvos no arquivo de trajetória com as opções iprint e iprintxyz:
julia> out = md(sys,Options(dt=0.1,iprint=1,iprintxyz=5))O número total de passos é controlado pelo parâmetro nsteps.
A variável out que sai da simulação é uma matriz com as energias e a temperatura em cada passo da simulação. É provável que a simulação "exploda" com passos de tempo grandes. Para visualizar esse processo, podemos fazer:
julia> using Plots
julia> plot(
out,ylim=[-100,100],
label=["Potential" "Kinetic" "Total" "Temperature"],
xlabel="step"
)E obteremos um gráfico semelhante a:
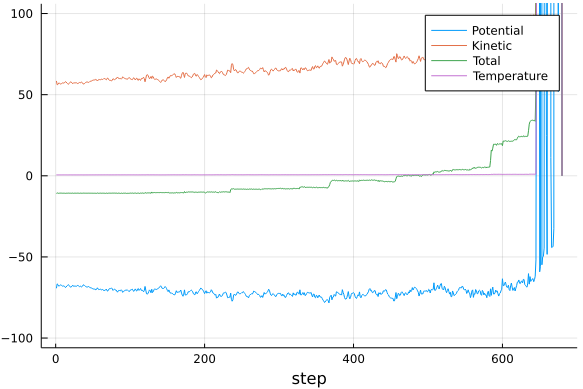
Para passos de tempo menores a simulação deve conseguir chegar até o fim. Podemos ver o resultado novamente, e deve ser algo semelhante a:
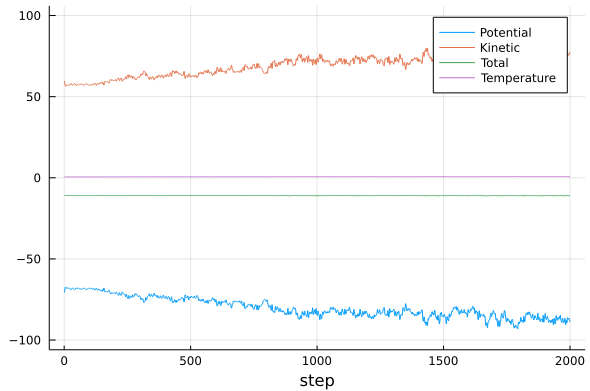
Observe e tente entender as amplitudes das oscilações das energias cinética e potencial, e as amplitudes das oscilações da energia total. A que se devem cada uma das oscilações? Veja como essas oscilações dependem do passo de integração.
3.2. Visualização da trajetória
Por fim, abra a trajetória usando VMD. Não é necessário sair da seção do Julia. Ao pressionar ; (ponto e vírgula) aparecerá um prompt shell>, a partir do qual é possível executar o VMD, se este estiver instalado corretamente e disponível no path:
shell> vmd traj.xyzDentro do VMD, escolha a representação VDW, em
Graphics -> Representations -> Drawing Method -> VDWe dê o comando
vmd> pbc set { 100. 100. 100. } -allpara indicar a periodicidade do sistema. Para representar explicitamente o sistema periódico, escolha +X;+Y;-X;-Y em
Graphics -> Representations -> PeriodicPara sair do VMD use o comando exit, e para voltar ao prompt do Julia a partir do shell>, use backspace.
3.3. Código completo resumido
using FundamentosDMC, Plots
sys = System(n=100)
minimize!(sys)
out = md(sys,Options(dt=0.05))
plot(out,ylim=[-100,100],
label=["Potential" "Kinetic" "Total" "Temperature"],
xlabel="step"
)O arquivo traj.xyz é gerado e pode ser visualizado no VMD.